La
retinosis pigmentaria es la enfermedad hereditaria de la retina más frecuente. Es una
enfermedad genética en la que el paciente pierde lenta y progresivamente la visión. Es considerada una
enfermedad rara que sufre 1 entre 4000 personas y afecta más a los hombres, en una proporción del 60 por ciento, frente al 40 por ciento de mujeres.
La retinosis pigmentaria no es una única enfermedad sino un conjunto de enfermedades de
origen degenerativo con multitud de mutaciones identificadas y asociadas al desarrollo de unos signos y síntomas comunes. El grado de afectación puede ser muy diferente entre pacientes, así como la velocidad a la que avanza. El componente hereditario
también varía y hasta en un 50 por ciento de los pacientes no tienen antecedente familiar conocido. Las formas recesivas y aquellas con comienzo temprano de los síntomas tienen peor pronóstico.
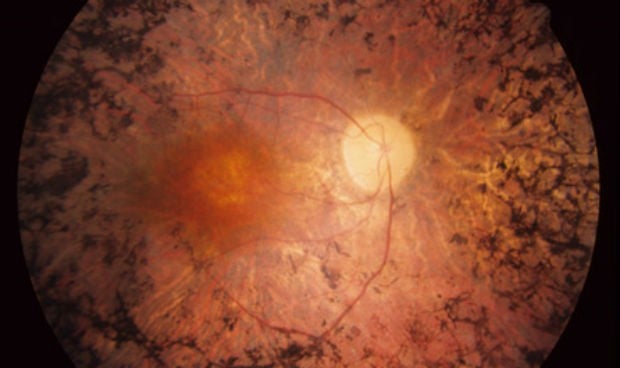
Esta dolencia se caracteriza por la afectación progresiva de los fotorreceptores: inicialmente de los bastones, los encargados de la visión nocturna y de la mayoría del campo visual. En fases avanzadas también afecta a los conos, los encargados de la visión central y de la percepción de los colores, explican desde la
Unidad de
Oftalmología HLA Montpellier.
También conocida como retinitis pigmentosa,
esta enfermedad crónica puede llevar a la ceguera. Los síntomas suelen aparecer en la adolescencia o en los primeros 30 años de vida. El primer síntoma suele ser la ceguera nocturna o dificultad de ver con baja iluminación por una mala adaptación a la oscuridad. Otros síntomas que aparecen conforme avanza la enfermedad son la
reducción concéntrica del campo visual (visión en túnel), disminución de la sensibilidad al contraste y disminución progresiva de la agudeza visual. Cuando la enfermedad está muy avanzada, no se perciben con claridad los colores y aparecen deslumbramientos frecuentes y fotopsias (luces o flashes). Además, puede ocasionar la aparición de problemas derivados como edema macular cistoide y cataratas.
¿Cómo se diagnostica esta enfermedad?
Para diagnosticar la enfermedad, el oftalmólogo realiza un estudio del fondo de ojo con dilatación de la pupila, donde se encuentran las lesiones pigmentadas típicas llamadas espículas. “Otras pruebas que se realizan y que nos sirven para el diagnóstico y para el seguimiento de la enfermedad son la agudeza visual, el campo visual, un electrorretinograma y pruebas de adaptación a la oscuridad”, matizan los especialistas de HLA Montpellier. Si se considera necesario se puede realizar un estudio genético para determinar qué mutación tiene el gen afectado, ya que existe la posibilidad de transmitir la enfermedad a su descendencia.
En cuanto al tratamiento, por el momento no existe una cura estándar y efectiva, si bien hay muchas líneas de investigación abiertas al respecto. Al tratarse de un amplio grupo de enfermedades no es fácil encontrar una única solución.
Para un tipo concreto de retinosis pigmentaria, la que afecta al gen RPE65, la UE aprobó recientemente un medicamento que introduce en los fotorreceptores enfermos copias del gen sin la mutación mediante terapia génica. También se han desarrollado implantes electrónicos retinianos que pueden proporcionar algo de visión en estadios muy avanzados de la enfermedad.
Una vez realizado el diagnóstico, la labor del oftalmólogo en el seguimiento y en la educación del paciente y su familia sobre su enfermedad es muy importante. Los pacientes con retinitis pigmentaria afrontan una pérdida progresiva de visión sin saber en cuánto tiempo se puede producir esa pérdida, por lo que deberán poco a poco adaptarse a esta situación. El apoyo psicológico, así como las asociaciones de pacientes con esta patología pueden ser útiles.
Las informaciones publicadas en Redacción Médica contienen afirmaciones, datos y declaraciones procedentes de instituciones oficiales y profesionales sanitarios. No obstante, ante cualquier duda relacionada con su salud, consulte con su especialista sanitario correspondiente.






 Andalucía
Andalucía  Cataluña
Cataluña  Madrid
Madrid  C. Valenciana
C. Valenciana  Galicia
Galicia  Castilla y León
Castilla y León  País Vasco
País Vasco  Canarias
Canarias  C-La Mancha
C-La Mancha  Murcia
Murcia  Aragón
Aragón  Extremadura
Extremadura  Asturias
Asturias  Baleares
Baleares  Navarra
Navarra  Cantabria
Cantabria  La Rioja
La Rioja 
